Proteinfaltung
Dies ist ein Vorschlag, um das Problem der Proteinfaltung mit Hilfe eines Neuronalen Netzwerks zu lösen.
Unser Verfahren
Das gesamte Verfahren wird hier vollständig beschrieben, damit jeder es nachmachen und weiter verbessern kann. Dieses Verfahren ist eine vollständig eigene Entwicklung von Kroll-Software und basiert auf keinen anderen Ideen. Bitte zitieren sie uns, wenn Sie dieses Verfahren verwenden.
Das Verfahren macht eine Voraussage über den (euklidischen) Abstand zweier Atome in einem Protein. Das funktioniert erstaunlich gut und führt in vielen Fällen zu sehr guten Faltungen.
Motivation
Um den Abstand lernen zu können, übergeben wir Paare von zwei Atomen im Kontext ihrer weiteren benachbarten Amino-Sequenz. Insofern handelt es sich um ein N-Gram Verfahren (N=27). Die Reihenfolge der Sequenz ist wichtig und soll für spätere Netzwerk-Schichten kodiert werden. Zusätzlich wird die Länge der gesamten Protein-Sequenz sowie der Sequenz-Abstand beider Atome übergeben.
Daten
Die verwendeten Trainings-Daten stammen aus der öffentlich zugänglichen Protein Datenbank (PDB). Die gepackten Daten sind 42 GB gross. Zur schnelleren Abfrage haben wir die Daten in eine lokale PostgreSQL Datenbank eingelesen.
Implementierung
Das Neuronale Netzwerk nutzt Keras / Tensorflow (oder ein anderes Backend). Wir programmieren in C# und verwenden eine modifizierte Version von Keras.NET. Alle Codebeispiele sind daher in C#, lassen sich aber leicht in andere Sprachen adaptieren. Unsere Arbeitsumgebung und die Screenshots sind eine SummerGUI-Anwendung unter Mono/Ubuntu, aber das spielt für das Verfahren keine Rolle.
Netzwerk Architektur / Topologie
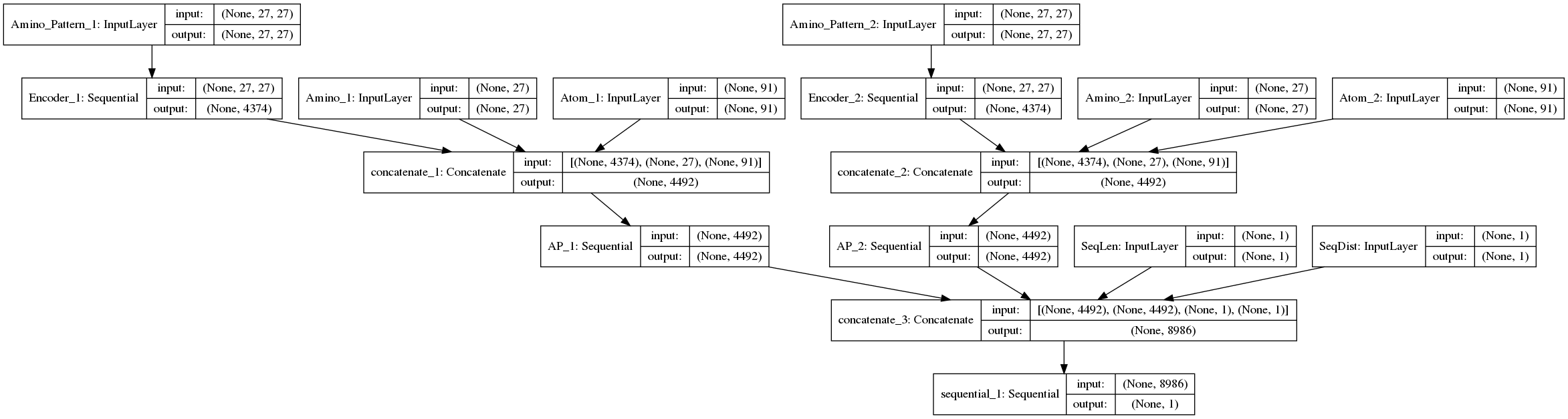
Dieses Modell besteht aus mehreren Sub-Modellen. Wir verwenden die Keras Functional API, um dieses komplexe Modell zusammenzusetzen.
private BaseModel BuildModel()
{
int aminoLen = Enum.GetValues(typeof(PDB.Model.Amino.Codes)).Length; // 27
int atomLen = AtomNames.Count; // 91
int timesteps = aminoLen; // 27
var aminoPatternShape = new Shape(timesteps, aminoLen);
var aminoShape = new Shape(aminoLen);
var atomShape = new Shape(atomLen);
var valueShape = new Shape(1);
float dropOut = 0.1f;
bool useBias = false;
int df = 4;
// Inputs
var inputAminoPattern1 = new Input(shape: aminoPatternShape, name: "Amino_Pattern_1");
var inputAminoPattern2 = new Input(shape: aminoPatternShape, name: "Amino_Pattern_2");
var inputAmino1 = new Input(shape: aminoShape, name: "Amino_1");
var inputAtom1 = new Input(shape: atomShape, name: "Atom_1");
var inputAmino2 = new Input(shape: aminoShape, name: "Amino_2");
var inputAtom2 = new Input(shape: atomShape, name: "Atom_2");
var inputSeqLen = new Input(shape: valueShape, name: "SeqLen");
var inputSeqDist = new Input(shape: valueShape, name: "SeqDist");
// my modified Keras.NET allows null for 'None'
var noiseShape = new Shape(null, 1, aminoLen);
// 1. Amino_Pattern_1
Sequential encoder1 = new Sequential("Encoder_1");
encoder1.Add(new LSTM(timesteps, input_shape: aminoPatternShape, return_sequences: true));
encoder1.Add(new Dropout(dropOut, noise_shape: noiseShape));
encoder1.Add(new Conv1D(filters: timesteps * timesteps * 2, kernel_size: 9, strides: 1));
encoder1.Add(new GlobalMaxPooling1D());
var outputAminoPattern1 = encoder1.Call(inputAminoPattern1);
encoder1.Summary();
// 2. Amino_Pattern_2
Sequential encoder2 = new Sequential("Encoder_2");
encoder2.Add(new LSTM(timesteps, input_shape: aminoPatternShape, return_sequences: true));
encoder2.Add(new Dropout(dropOut, noise_shape: noiseShape));
encoder2.Add(new Conv1D(filters: timesteps * timesteps * 2, kernel_size: 9, strides: 1));
encoder2.Add(new GlobalMaxPooling1D());
var outputAminoPattern2 = encoder2.Call(inputAminoPattern2);
var mergeA1 = new Concatenate(outputAminoPattern1, inputAmino1, inputAtom1);
var mergeA2 = new Concatenate(outputAminoPattern2, inputAmino2, inputAtom2);
Sequential decodeAP1 = new Sequential("AP_1");
decodeAP1.Add(new LeakyReLU());
decodeAP1.Add(new Dense(timesteps * timesteps * df, use_bias: useBias, kernel_initializer: "random_normal"));
decodeAP1.Add(new LeakyReLU());
decodeAP1.Add(new Dense(timesteps * timesteps * df, use_bias: useBias, kernel_initializer: "random_normal"));
var outAP1 = decodeAP1.Call(mergeA1);
Sequential decodeAP2 = new Sequential("AP_2");
decodeAP2.Add(new LeakyReLU());
decodeAP2.Add(new Dense(timesteps * timesteps * df, use_bias: useBias, kernel_initializer: "random_normal"));
decodeAP2.Add(new LeakyReLU());
decodeAP2.Add(new Dense(timesteps * timesteps * df, use_bias: useBias, kernel_initializer: "random_normal"));
var outAP2 = decodeAP2.Call(mergeA2);
var mergeLayer = new Concatenate(outAP1, outAP2, inputSeqLen, inputSeqDist);
Sequential decoder = new Sequential();
decoder.Add(new LeakyReLU());
decoder.Add(new Dense(units: 1460 * df, use_bias: useBias, kernel_initializer: "random_normal"));
decoder.Add(new LeakyReLU());
decoder.Add(new Dense(units: 730 * df, use_bias: useBias, kernel_initializer: "random_normal"));
decoder.Add(new LeakyReLU());
decoder.Add(new Dense(units: 730 * df, use_bias: useBias, kernel_initializer: "random_normal"));
decoder.Add(new Dense(units: 1, use_bias: useBias));
var output = decoder.Call(mergeLayer);
var model = new Model(inputs: new BaseLayer[] { inputAminoPattern1, inputAminoPattern2, inputAmino1, inputAmino2, inputAtom1, inputAtom2, inputSeqLen, inputSeqDist }, outputs: new BaseLayer[] { output });
var optimizer = new RMSprop(lr: 1e-4f);
model.Compile(optimizer: optimizer,
loss: "mse",
metrics: new string[] { "mean_absolute_percentage_error" });
model.Summary();
model.Plot(show_layer_names: true);
return model;
}
Eingabewerte
Das Model besitzt mehrere Input Objekte für die verschiedenen Eingabewerte.
Amino Muster (2x)
Eine quadratische 27x27 Matrix aus One-Hot kodierten Werten von Aminosäuren. Horizontal die Sequenz-Position, vertikal der kodierte Index (jeder Wert ist 1 oder 0). Wie bei N-Gram Verfahren üblich, erhalten führende und endende undefinierte Positionen einen eigenen kodierten Wert, der nicht 0 ist. Die aktuelle Aminosäure des Atoms liegt in der Mitte bei Index 13.
Amino (2x)
Eine 1x27 Matrix mit einem One-Hot kodierten Wert der aktuellen Aminosäure des Atoms.
Atom (2x)
Eine 1x91 Matrix mit einem One-Hot kodierten Wert aller vorkommenden Atom-Bezeichner (z.B."CA", "1HB", ..).
Sequenz Abstände
Die gesamte Amino-Sequenz Länge (Anzahl der Residuen) sowie die Amino-Distanz der beiden Atome.
Ausgabewert
Das Model liefert einen einzigen Ausgabewert, das ist der (euklidische) Abstand der beiden Atome.
Normalisierung der Werte
Damit die Gewichtungen im Neuronalen Netz nicht eskalieren, ist es wichtig, die Eingabewerte zu skalieren, sodass sie ungefähr zwischen 0 und 1 liegen.
- Ground-Truth Abstände und Ausgabewert werden mit dem Faktor 10 skaliert.
- Sequenz-Längen und Abstände werden durch 100 geteilt.
Training
In der gegenwärtigen Version beschränken wir uns auf das jeweils erste Modell in einer PDB-Datei und trainieren Proteine mit 500 bis 3000 Atomen (das sind ca. 90% der Proteine in der PDB). Wir verzichten auf die ganz kleinen und ganz grossen Proteine, um schneller zum Ergebnis zu kommen. Auf einer einzelnen GPU benötigt der Trainingslauf ca. 2 Wochen über die gesamte PDB, bis das Netzwerk die Daten gut angenähert hat.
Vorwärts gerichtetes Einlesen der Daten
Wir lesen die Daten ausschliesslich vorwärts gerichtet ein und fragen sie später bei den Predictions auch nur in dieser Richtung ab, um nicht jeden Abstand doppelt zu trainieren. For Atom i=0 to n .. for Atom k=i+1 to n ..
Custom Batch-Train
Wegen der riesigen Datenmenge verwenden wir eine Custom Training-Loop mit der Keras-Funktion train_on_batch(). Entscheidend dabei ist die Batch-Size. Wir trainieren in einem Lernschritt 51.200 zufällige Atom-Beziehungen aus jeweils 5 zufälligen Proteinen (das sind weniger als 1% aller Atom-Beziehungen eines Proteins je Lernschritt) und partitionieren diese auf Mini-Batches von 512 Werten.
Ground-Truth
Target data beim batch-train ist eine Liste der skalierten (euklidischen) Ground-Truth Abstände beider Atome.
Optimierer
Wir verwenden den RMSprop Optimierer. Wir beginnen mit einer Lernrate von 1e-4 und setzen diese später auf 2.5e-5 herab.
Drop-Out
Anfangs kann das Netz nur mit einem Drop-Out lernen. Später kann der Drop-Out auf 0 gesetzt werden, um bessere Ergebnisse zu erzielen.
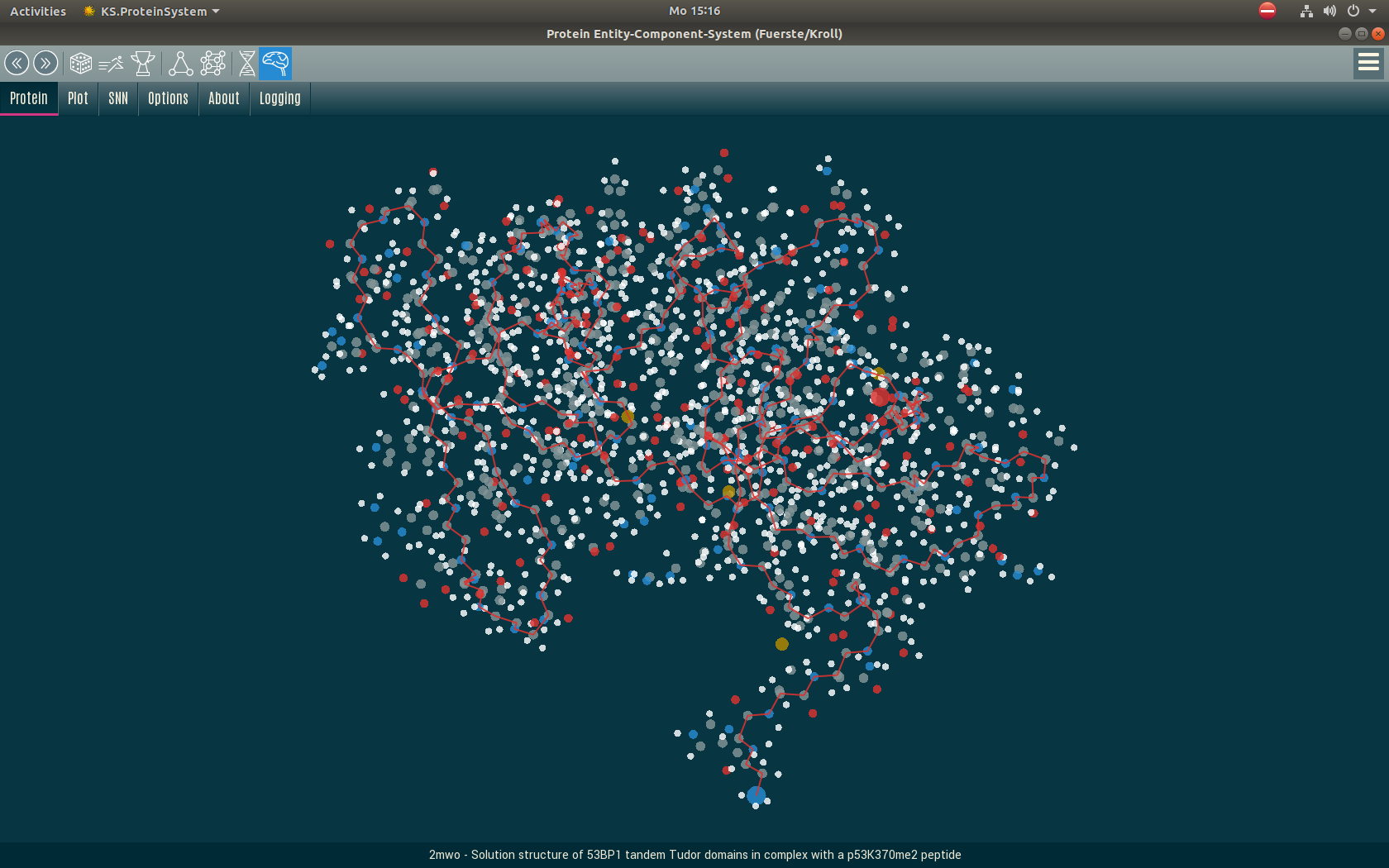
Partikelsystem
Zur Faltung eines Proteins sind bis zu 5 Mio. Network-Predictions erforderlich, das geht in wenigen Minuten. Für die eigentliche Faltung aus den Atomabständen verwenden wir ein Partikelsystem, welches als Entity-Component-System (ECS) implementiert ist.
Validierung
Wir validieren unsere Faltungen mit der freien Software MaxCluster. Damit berechnen wir einen Global distance test (GDT), TM-Score, RMSD und andere Metriken.
Credits
Citation needed.
Unterstützen Sie diese Forschung
Wenn Ihnen diese Forschung gefällt, freuen wir uns auf Ihre Unterstützung. Bitte kontaktieren Sie uns.