Protein Folding
This is a proposal to solve the problem of protein folding with the help of a neural network.
Our Method
The entire process is fully described here for anyone to copy and improve on. This process is a completely in-house development by Kroll-Software and is not based on any other ideas. Please quote us if you use this method.
The procedure makes a prediction about the (Euclidean) distance between two atoms in a protein. This works amazingly well and in many cases leads to very good folds.
Motivation
In order to be able to learn the distance, we pass pairs of two atoms in the context of their further neighboring amino sequence. In this respect, it is an N-Gram procedure (N=27). The order of the sequence is important and should be coded for later network layers. In addition, the length of the entire protein sequence and the sequence distance between the two atoms are transferred.
Data
The training data used come from the publicly accessible Protein Database (PDB). The packed data is 42 GB. For a faster query we have read the data into a local PostgreSQL database.
Implementation
The neural network uses Keras / Tensorflow (or another backend). We program in C# and use a modified version of Keras.NET. All code examples are therefore in C#, but can easily be adapted to other languages. Our working environment and the screenshots are a SummerGUI application under Mono/Ubuntu, but that does not matter for the process.
Network Architecture / Topology
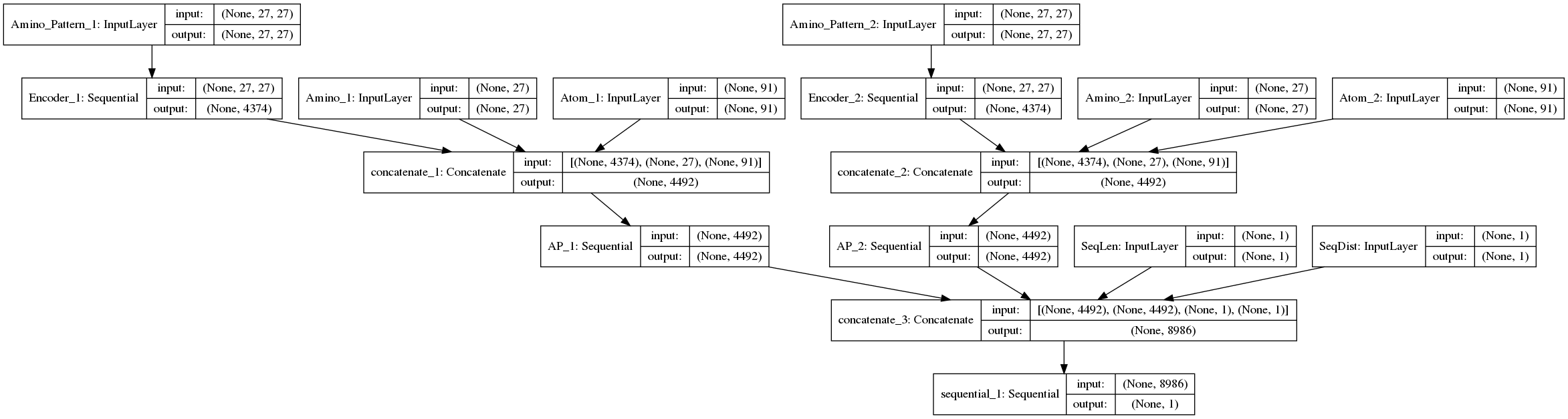
This model consists of several sub-models. We use the Keras Functional API to put this complex model together.
private BaseModel BuildModel()
{
int aminoLen = Enum.GetValues(typeof(PDB.Model.Amino.Codes)).Length; // 27
int atomLen = AtomNames.Count; // 91
int timesteps = aminoLen; // 27
var aminoPatternShape = new Shape(timesteps, aminoLen);
var aminoShape = new Shape(aminoLen);
var atomShape = new Shape(atomLen);
var valueShape = new Shape(1);
float dropOut = 0.1f;
bool useBias = false;
int df = 4;
// Inputs
var inputAminoPattern1 = new Input(shape: aminoPatternShape, name: "Amino_Pattern_1");
var inputAminoPattern2 = new Input(shape: aminoPatternShape, name: "Amino_Pattern_2");
var inputAmino1 = new Input(shape: aminoShape, name: "Amino_1");
var inputAtom1 = new Input(shape: atomShape, name: "Atom_1");
var inputAmino2 = new Input(shape: aminoShape, name: "Amino_2");
var inputAtom2 = new Input(shape: atomShape, name: "Atom_2");
var inputSeqLen = new Input(shape: valueShape, name: "SeqLen");
var inputSeqDist = new Input(shape: valueShape, name: "SeqDist");
// my modified Keras.NET allows null for 'None'
var noiseShape = new Shape(null, 1, aminoLen);
// 1. Amino_Pattern_1
Sequential encoder1 = new Sequential("Encoder_1");
encoder1.Add(new LSTM(timesteps, input_shape: aminoPatternShape, return_sequences: true));
encoder1.Add(new Dropout(dropOut, noise_shape: noiseShape));
encoder1.Add(new Conv1D(filters: timesteps * timesteps * 2, kernel_size: 9, strides: 1));
encoder1.Add(new GlobalMaxPooling1D());
var outputAminoPattern1 = encoder1.Call(inputAminoPattern1);
encoder1.Summary();
// 2. Amino_Pattern_2
Sequential encoder2 = new Sequential("Encoder_2");
encoder2.Add(new LSTM(timesteps, input_shape: aminoPatternShape, return_sequences: true));
encoder2.Add(new Dropout(dropOut, noise_shape: noiseShape));
encoder2.Add(new Conv1D(filters: timesteps * timesteps * 2, kernel_size: 9, strides: 1));
encoder2.Add(new GlobalMaxPooling1D());
var outputAminoPattern2 = encoder2.Call(inputAminoPattern2);
var mergeA1 = new Concatenate(outputAminoPattern1, inputAmino1, inputAtom1);
var mergeA2 = new Concatenate(outputAminoPattern2, inputAmino2, inputAtom2);
Sequential decodeAP1 = new Sequential("AP_1");
decodeAP1.Add(new LeakyReLU());
decodeAP1.Add(new Dense(timesteps * timesteps * df, use_bias: useBias, kernel_initializer: "random_normal"));
decodeAP1.Add(new LeakyReLU());
decodeAP1.Add(new Dense(timesteps * timesteps * df, use_bias: useBias, kernel_initializer: "random_normal"));
var outAP1 = decodeAP1.Call(mergeA1);
Sequential decodeAP2 = new Sequential("AP_2");
decodeAP2.Add(new LeakyReLU());
decodeAP2.Add(new Dense(timesteps * timesteps * df, use_bias: useBias, kernel_initializer: "random_normal"));
decodeAP2.Add(new LeakyReLU());
decodeAP2.Add(new Dense(timesteps * timesteps * df, use_bias: useBias, kernel_initializer: "random_normal"));
var outAP2 = decodeAP2.Call(mergeA2);
var mergeLayer = new Concatenate(outAP1, outAP2, inputSeqLen, inputSeqDist);
Sequential decoder = new Sequential();
decoder.Add(new LeakyReLU());
decoder.Add(new Dense(units: 1460 * df, use_bias: useBias, kernel_initializer: "random_normal"));
decoder.Add(new LeakyReLU());
decoder.Add(new Dense(units: 730 * df, use_bias: useBias, kernel_initializer: "random_normal"));
decoder.Add(new LeakyReLU());
decoder.Add(new Dense(units: 730 * df, use_bias: useBias, kernel_initializer: "random_normal"));
decoder.Add(new Dense(units: 1, use_bias: useBias));
var output = decoder.Call(mergeLayer);
var model = new Model(inputs: new BaseLayer[] { inputAminoPattern1, inputAminoPattern2, inputAmino1, inputAmino2, inputAtom1, inputAtom2, inputSeqLen, inputSeqDist }, outputs: new BaseLayer[] { output });
var optimizer = new RMSprop(lr: 1e-4f);
model.Compile(optimizer: optimizer,
loss: "mse",
metrics: new string[] { "mean_absolute_percentage_error" });
model.Summary();
model.Plot(show_layer_names: true);
return model;
}
Input Values
The model has several input objects for the various input values.
Amino Pattern (2x)
A 27x27 square matrix of one-hot coded values of amino acids. The sequence position horizontally, the coded index vertically (each value is 1 or 0). As usual with N-Gram procedures, leading and ending undefined positions receive their own coded value that is not 0. The current amino acid of the atom is in the middle at index 13.
Amino (2x)
A 1x27 matrix with a one-hot coded value of the current amino acid of the atom.
Atom (2x)
A 1x91 matrix with a one-hot coded value of all occurring atom identifiers (e.g. "CA", "1HB", ..).
Sequence Distances
The total amino sequence length (number of residues) and the amino distance between the two atoms.
Output Value
The model delivers a single output value, that is the (Euclidean) distance between the two atoms.
Normalization of Values
So that the weights in the neural network do not escalate, it is important to scale the input values so that they are approximately between 0 and 1.
- Ground truth distances and output value are scaled with a factor of 10.
- Sequence lengths and distances are divided by 100.
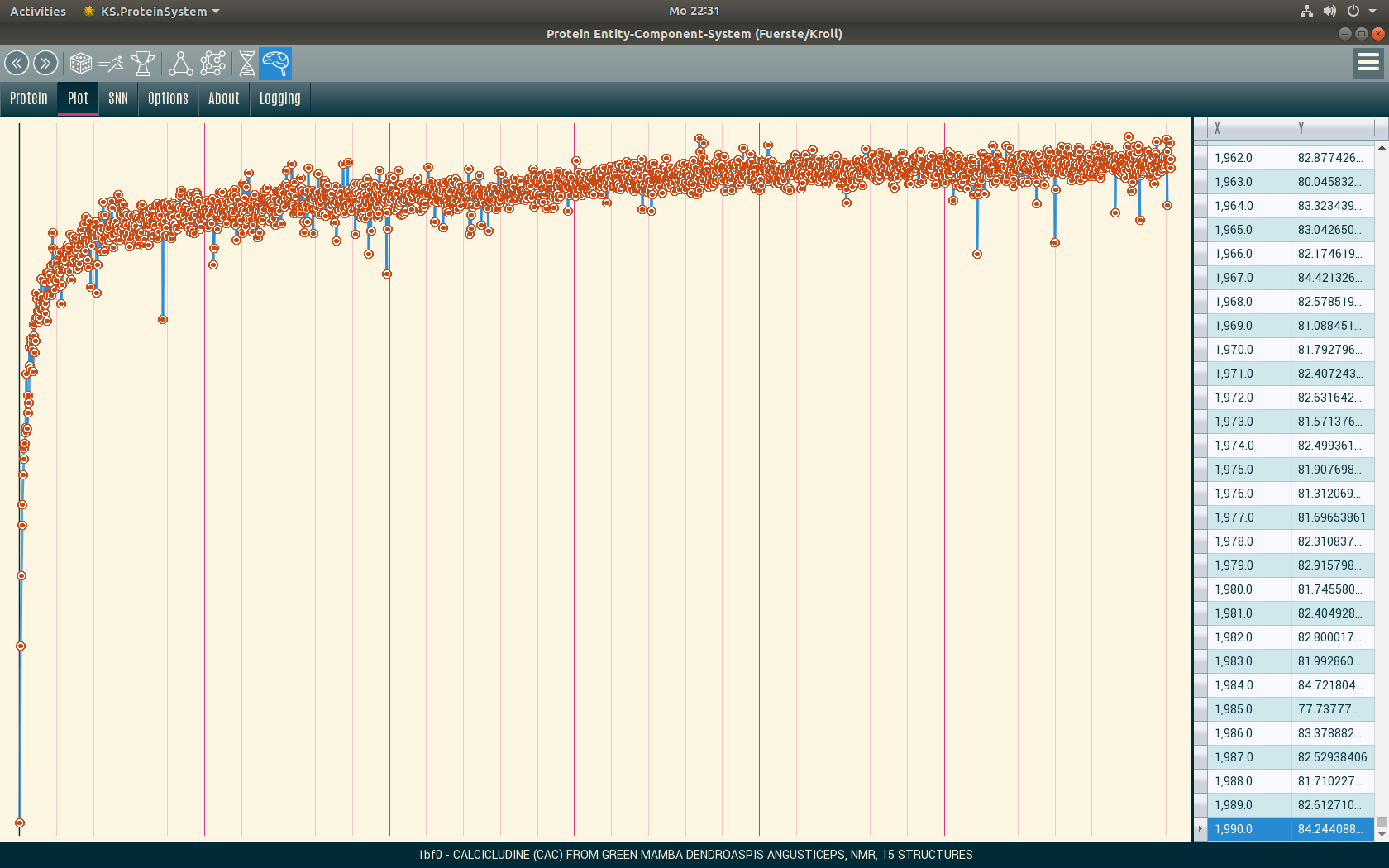
Training
In the current version we limit ourselves to the first model in a PDB file and train proteins with 500 to 3000 atoms (that is approx. 90% of the proteins in the PDB). We do without the very small and very large proteins in order to get results faster. On a single GPU, the training run takes about 2 weeks over the entire PDB until the network has approximated the data well.
Forward processing of the data
We only read the data in a forward-facing direction and later only query them in this direction during the predictions, so as not to train every distance twice. For Atom i=0 to n .. for Atom k=i+1 to n ..
Custom Batch-Train
Because of the huge amount of data, we use a custom training loop with the Keras function train_on_batch(). The decisive factor here is the Batch-Size. In one learning step we train 51,200 random atomic relationships from 5 random proteins each (that is less than 1% of all atomic relationships of a protein per learning step) and partition them into mini-batches of 512 values.
Ground-Truth
Target data for the batch train is a list of the scaled (Euclidean) ground truth distances between the two atoms.
Optimizer
We use the RMSprop optimizer. We start with a learning rate of 1e-4 and later reduce it to 2.5e-5.
Drop-Out
Initially, the network can only learn with a drop-out. The drop-out can later be set to 0 for better results.
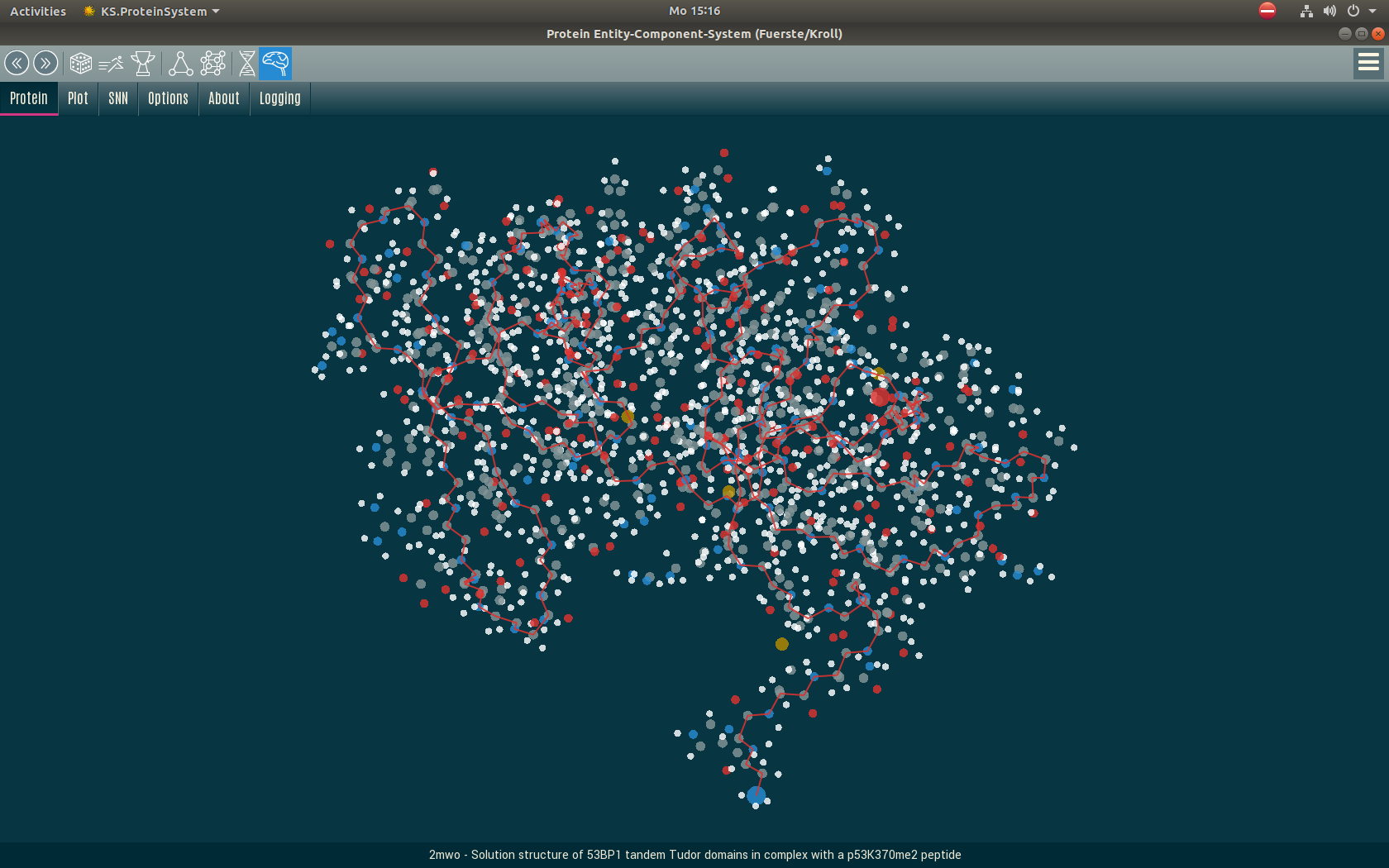
Particle System
Up to 5 million network predictions are required to fold a protein, which can be done in a few minutes. For the actual convolution from the atomic distances, we use a particle system that is implemented as an Entity Component System (ECS).
Validation
We validate our folds with the free software MaxCluster. We use it to calculate a Global distance test (GDT), TM-Score, RMSD and other metrics.
Credits
Citation needed.
Support this Research
If you enjoy this research, we look forward to your support. Please contact us.